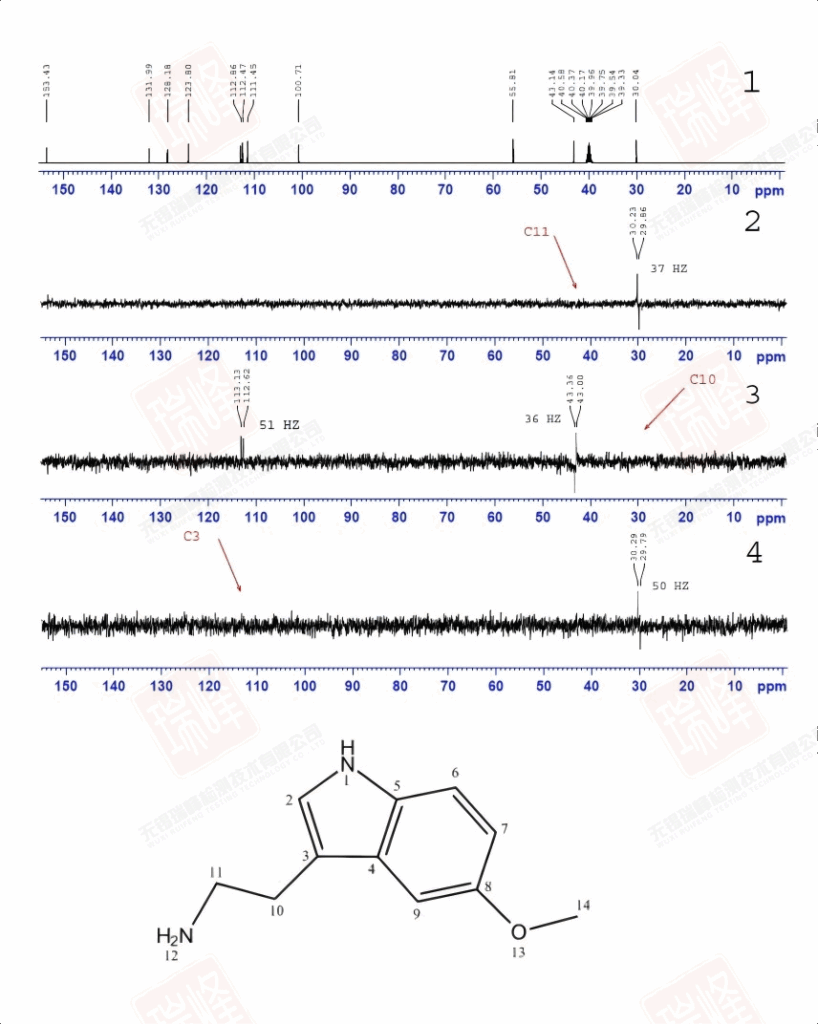
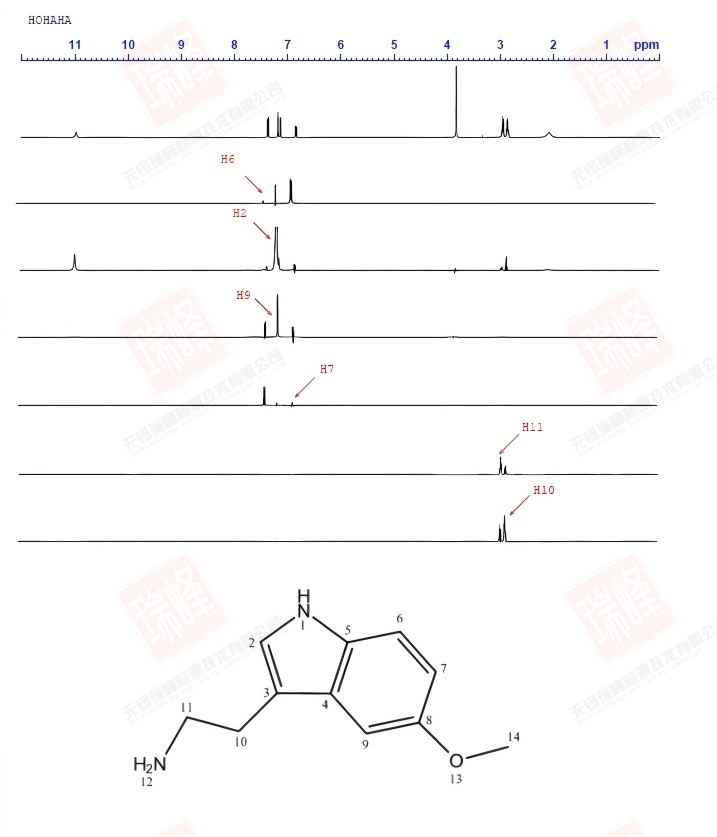
被称之为SELINA的选择性1D-INADQUATE实验通过软脉冲照射特定核(其实照射的是某一13C的两个13C卫星峰)来观察与之相连的13C信号。
图一为普通碳谱。
图二为照射C11后的谱图,与C11相连的C10呈现反相位双重峰,双重峰的中间位置正好是图一中C10的化学位移。
图三在照射C10后除了出现C11的反相位峰外,远处的C3亦被激发(为了使信号更明显,这里我没有校正C3的一级相位)。
图四中对C3的激发,使与之相连的C10得以呈现。
四张谱图均给出了CC耦合常数,连接关系一目了然。

被称之为SELINA的选择性1D-INADQUATE实验通过软脉冲照射特定核(其实照射的是某一13C的两个13C卫星峰)来观察与之相连的13C信号。
图一为普通碳谱。
图二为照射C11后的谱图,与C11相连的C10呈现反相位双重峰,双重峰的中间位置正好是图一中C10的化学位移。
图三在照射C10后除了出现C11的反相位峰外,远处的C3亦被激发(为了使信号更明显,这里我没有校正C3的一级相位)。
图四中对C3的激发,使与之相连的C10得以呈现。
四张谱图均给出了CC耦合常数,连接关系一目了然。
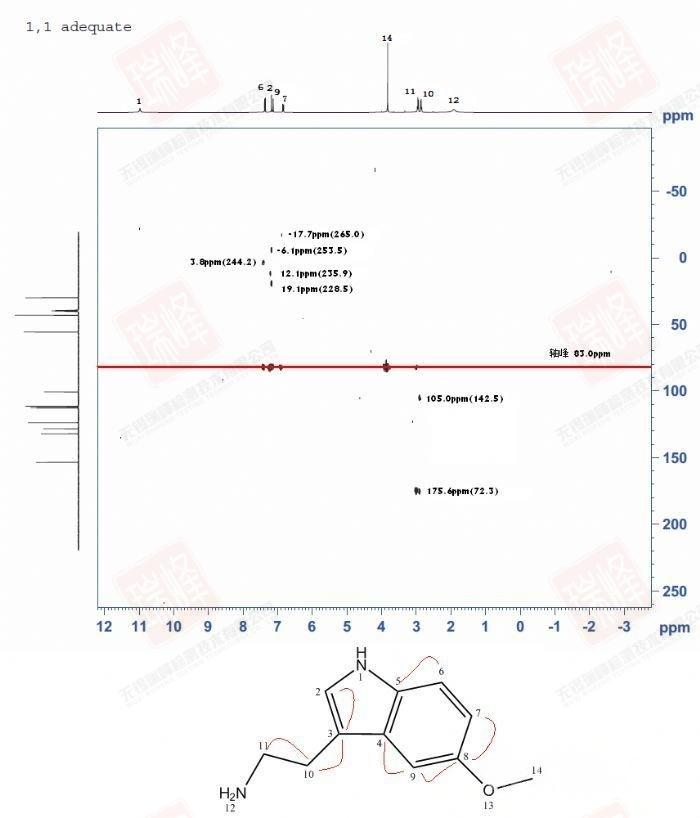
较2D-inadequate而言,1,1-adequate通过间接检测灵敏度更高的H核而使CC关系这一”不可能的任务”(inadequate)得以实现(adequate)。不过与inadequate不同,这一实验的局限在于只能给出CH-C这类结构的信号。
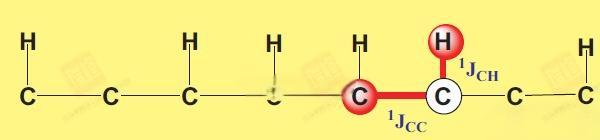
这一实验之所以会称为1,1-adequate,是因为所给出的信号是由J1HC传递到J1CC。为了克服之前所提到的局限性,不满足的核磁学家们开发出了一些传递更远的adequate,这个系列又称为n,n-adequate,以后有机会我会做这些谱放上来。
好在日常结构中三个季碳相邻的情况并不是很多,因此我们仍然能够借助于这一神奇的实验寻找CC连接的碎片来补全C骨架连接顺序的拼图。
由于这一实验的C维是双量子信号,因此纵向F1维的坐标并不能和C谱对应,而是相关CC化学位移的总和!下面我将着重介绍这一特殊谱图的解谱方法。
图中83 ppm处的轴峰(C的中心频率)是一种典型的系统干扰信号,因此我用红线划去。而不知道为什么,实验的F1维坐标是错的,为了和实际情况对应,这里我保留谱图的原貌,而在上图的括号中给出这个点应该在的坐标位置,大家可以通过计算实际点与谱图点之间的差值看到,这一坐标平移是遵循原谱各点化学位移的。F2维与对应的氢谱坐标一致。
上面给出了结构中各碳的化学位移值,以H10为例,在J1C10H10传递后H10分别与C11与C3存在J1HC关系。谱图中可以看到,H10在纵向F1维有两个相关点。而碳谱中相应化学位移如下:C10=29.6 ppm ; C11=42.7 ppm C3=112.4 ppm,稍加计算我们可以得到C10+C3=142.0 ppm ; C10+C11=72.3 ppm,这与两个相关点F1维坐标接近一致!不妨再计算下H9的关系,我们知道C9=100.3 ppm ; C4=127.8 ppm ; C8=153.0 ppm,那么根据结构上给出的连接关系,C9+C4=228.1 ppm ; C9+C8=253.3 ppm,这同样与H9在F1维的两个相关点坐标相同。限于时间关系,我不一一计算,大家感兴趣可以根据上面的方法自行推导——最终,我们得到如图结构中红线给出的连接关系,CC相关得到了归属!
需要注意的是,C6与C7相关没有在图中显现,这可能与我实验参数的选取有关,也可能是这一信号信噪比太弱导致,这也表明这类实验解谱需要像HMBC一样灵活应对,而不能过于死板。
总体而言,1,1-adequate在解决C-C信号灵敏度过低上有了很大的突破,但实际解谱中较为繁琐,F1维坐标不够直观。在和布鲁克工程师交流后,她向我推荐了另外一种更方便的实验,随后我将给出。
其实对判定结构而言,C核比H核更为重要,因为C是构成有机物的骨架,而H只是骨架上生出来的枝叶。遗憾的是,98.9%的12C由于自旋为0而不能被核磁检测到,而余下1.1%的13C因为旋磁比仅为H的1/4以及较长的弛豫时间导致核磁学家最终将H作为核磁实验中最重要的核。但C核绝没有被忽视。
之前已经提过,为了得到占C元素1.1%的13C信号,需要付出比H核多出数千倍的扫描时间。那么C-C相关信号呢?这个问题等价于分子中两个13C正好挨在一起的几率为多大——1.1%的平方。也就是说,10000个分子中只有一个分子某两个13C正好相邻,而要观察到这一个分子的CC相关,难度不仅在于如何“万里挑一”将这个很弱的信号采集出来,更在于如何“以一敌万”排除余下9999个分子大军的信号干扰。而描述这一问题的实验,科学家称之为INADEQUATE。
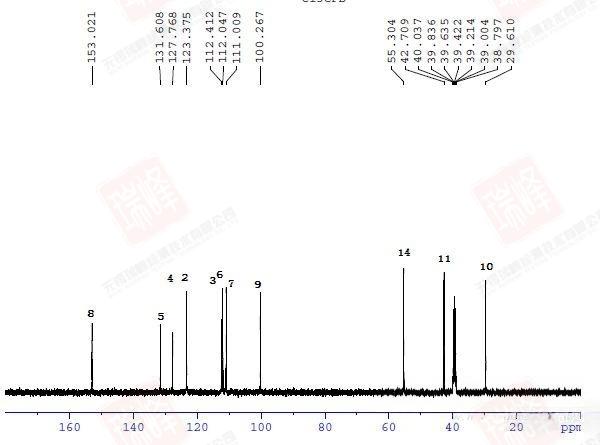
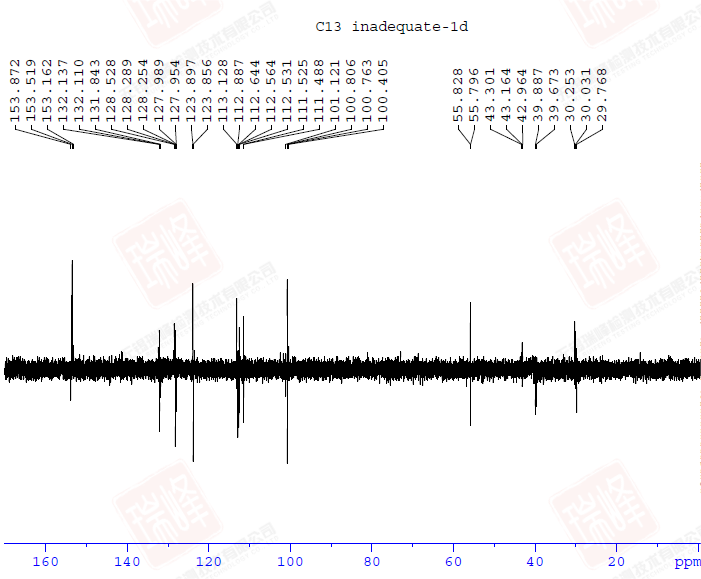
先给出这一结构的1D-INADEQUATE谱图。与2D相比,1D时间所花费的时间要少的多,但即使如此,这张谱图依然花费了我数小时的时间(还是在样品很浓的情况下)。由于脉冲演化的关系,1D-INADEQUATE实验中的信号常以反相位双重峰来呈现(其实稍微修改一下脉冲就可以实现相位同一化,但是我觉得这样反而不利于谱图解析),这是因为不管碳链多长,我们观察到的C-C相关通常就是一键相关(三个13C恰好相连的概率有多大?),而双重峰的波峰与波谷间距离是CC耦合常数,但遗憾的是,在1D-INADEQUATE中要分辨这一信号并不容易。
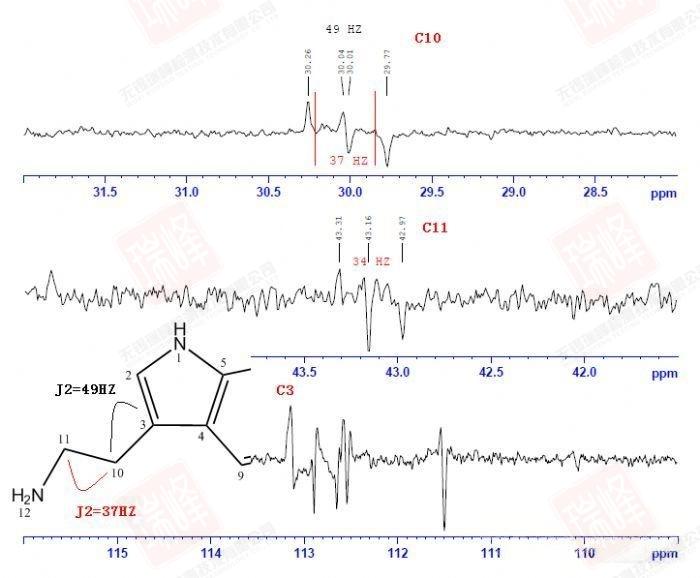
大家或许还记的之前在H归属中对于H10以及H11的归属曾经一度出现困难——HNMR中14N核四极矩造成的展宽以及H-N HMBC中相关信号的强弱都支持将高场H定为H11,但HMBC,COSY都明确给出了与之相反的结论。其实从C-C角度上来看这个问题会简单很多——C11仅受C10耦合,而C10却要受到C11和C3的耦合作用。图一中C10能明显读出49Hz的耦合作用,而红线标出的37Hz的反相位峰显得很弱,需要注意的是,在两个反相位双重峰中间位置出现的峰并不是三重峰,而是没有压制完全的13C单峰。图二给出的C11峰信号较弱,这可能是与之直接相连的14N所引起,34Hz的耦合值暗示着C10与C11的耦合关系。C3由于与C6与C7相互重叠,耦合变得难以辨认,使这一实验在解决C归属问题上不够严密,而我之后讲到的选择性1D-INADEQUATE将解决这个问题。
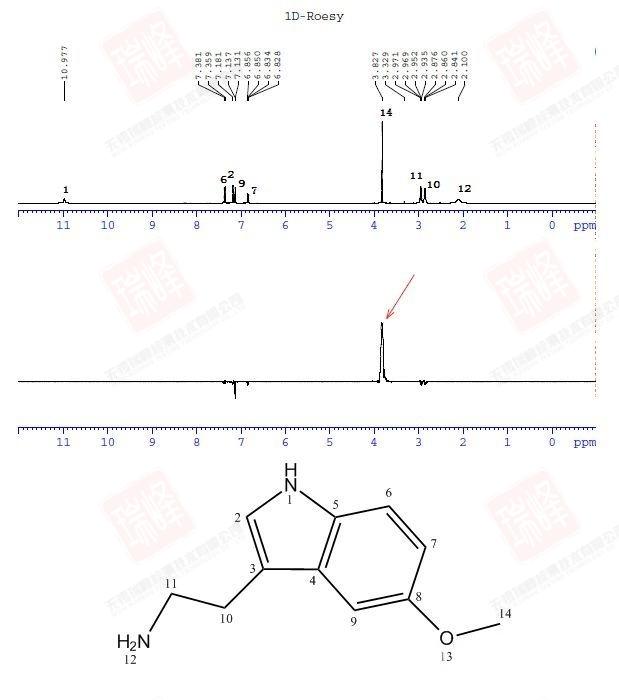
同1D-选择性NOESY相同,这一ROESY的1D版本能节省大量的实验时间或增加信噪比。需要注意的是,ROESY交叉峰在一维谱图上与照射峰呈现相反的相位。
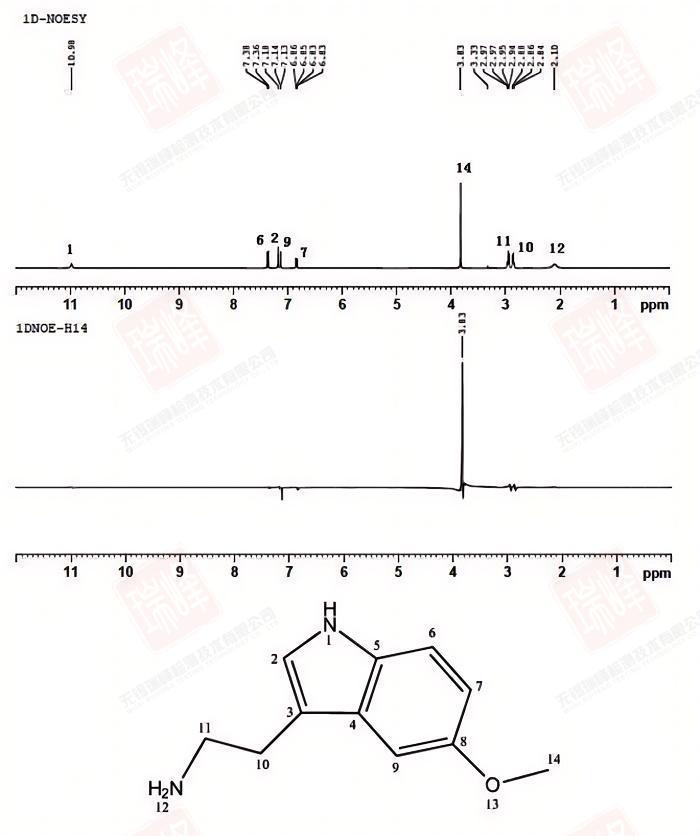
由于NOE信号增强很弱,因此NOESY常常需要扫描很长的时间(数小时到数天不等)。而通常情况是,实验人员并不需要得到整个结构的NOE关系,往往几个关键H之间的距离远近就足以对化合物结构做出一个准确的判断。在这种情况下,1D-选择性NOESY可以通过有目的地照射目标峰来快速得到需要的信息。
为了保持一致,这一实验的混合时间仍然定在1.0 s,选择性脉冲照射在了甲基的H上。在相同的扫描次数下,1D仅需要2D实验1/256的时间,却在H14上得到了和二维实验相同的结果。如果化合物浓度很低,那么节省下来的时间又可以成倍地用在增加扫描次数来提高信噪比上。事实上,在实际应用中,我更倾向于使用NOESY实验的1D版本。
需要注意的是,正负信号都有可能在空间上与照射峰接近,具体原因参见NOESY中正负峰的分析。
作为TOCSY的1D版本,这一实验能在很短的时间内,通过选择性脉冲,观测到与激发峰相关的H-H远程耦合信号。
如图所示,红色箭头所标示的是选择性激发的核,随着自旋锁定下接力相干传递,相关峰在谱图上以正相位显现出来。在混合时间相同的情况下,实验结果与2D实验一致。需要注意的是,当激发的H与相邻峰靠的比较近时,需要调节选择性脉冲的激发范围来避免“误伤”。
1D实验极大地缩短了实验时间(尤其在之后要提到的NOESY中),专一地提高了感兴趣信号的信噪比(与2D相比,NS可以设的很大),在不需要得到结构所有相关信号的情况下发挥着比其2D版本更为实用的作用。
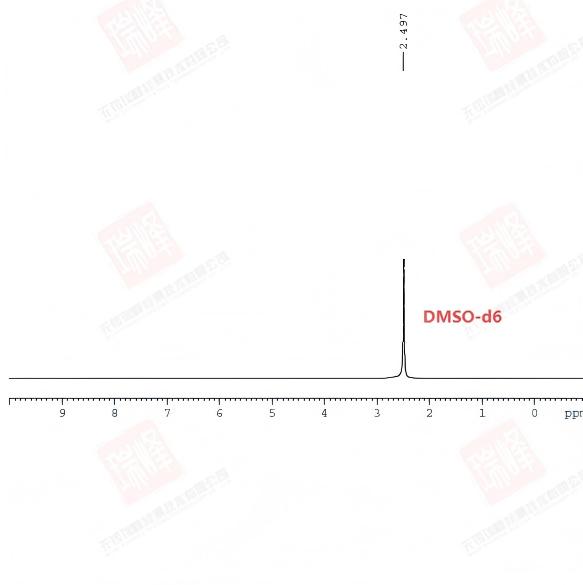
与14N相比,氘的核四极效应要小的多,从而能被核磁检测到。不过由于大部分核磁以D核作为锁场匀场观测核,因此采集D谱通常只能在脱锁状态下进行(有些专门做D谱的仪器则以F来锁场)。上图为我在脱锁状态下做的氘谱,2.5的单峰是氘代DMSO的甲基峰,核四极作用对峰型宽度影响不大。从图中可以看到,氘谱中D出峰的化学位移值与H几乎相同。
19F,31P以及29Si均为在核磁中做的比较多的杂核。与1H及13C一样,这类核在核磁共振检测中有一个很明显的优势——均为1/2核,因此谱图线型好,便于分析。通常杂核谱都对H核去耦而去除了H的耦合信息(类比于C13CPD),而通过化学位移来判断结构。由于我们的“单一化合物”并没有这三种核,因此我临时做了三张其他化合物的谱图来做一个简单说明。
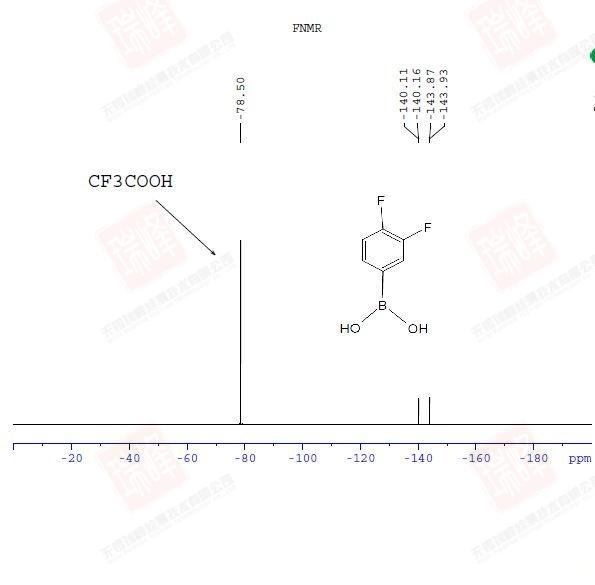
19F的核磁灵敏度与1H核相近,一般出峰范围在100—负300 ppm之间,通常用CF3Cl来标定0点,也可以用CF3COOH的-78.5 ppm来做标定。通常可以做F谱来判断化合物中是否含有F元素。
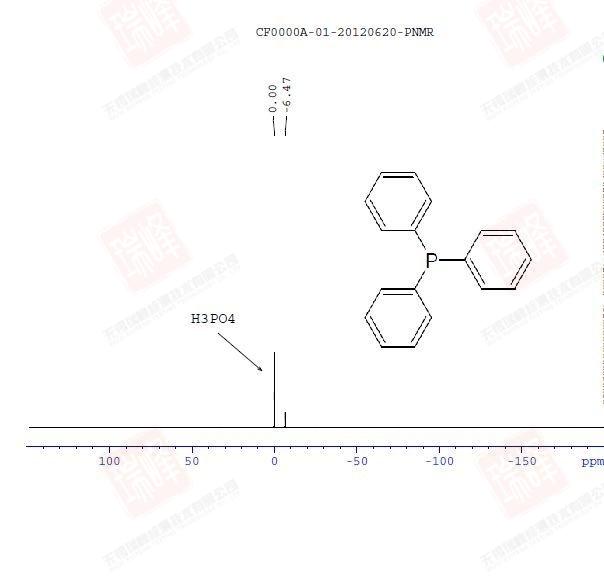
31P的灵敏度强于13C但弱于19F,但一般在较短的扫描时间内也能得到较为满意的谱图。谱图出峰集中在230—负200 ppm范围,通常用H3PO4标定0点。
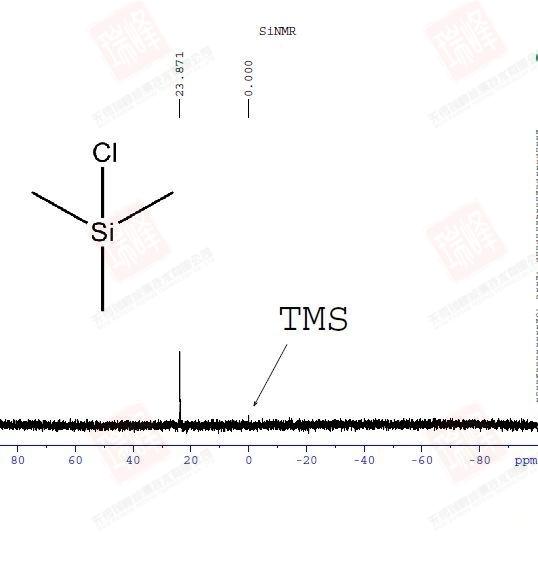
29Si灵敏度与13C接近,但是由于其旋磁比为负因此弛豫期间对H照射产生的NOE效应并不利于信号增强(甚至还会减弱)。核磁管玻璃中的29Si信号通常会对谱图产生干扰,因此需要核磁人员小心地进行基线处理扣除背景信号。29Si谱的出峰集中在100—负400,和大部分氢谱一样,TMS作为0点的标定。
总体而言,这三种1/2核是核磁常做的3种杂核,通常可以利用核磁来做一个快速的判断。而15N由于灵敏度太低通常需要同位素标记或富集后长时间扫描得到,之前提及的反向检测技术(见H-N HSQC/HMBC)在应对周围连有1H的15N核有特效。
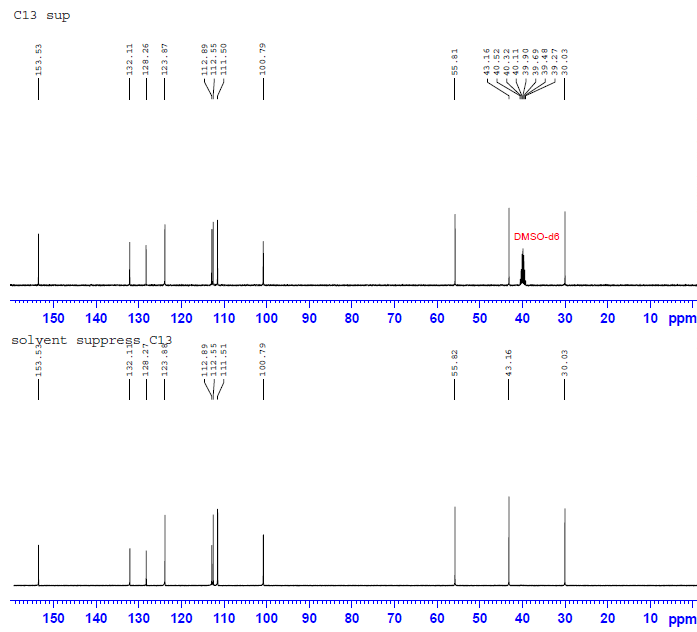
Bruker官方给出的标准脉冲中并没有碳溶剂峰压制实验,所以我自己编了一个碳溶剂压制脉冲,依然是拿我们的“标准化合物”下手。不过这一实验在我们高浓度的样品峰下除了能让谱图看起来更加直观,并没有任何效果。但是可以预计,在稀浓度的样品中,这一实验将会有它的用武之地。
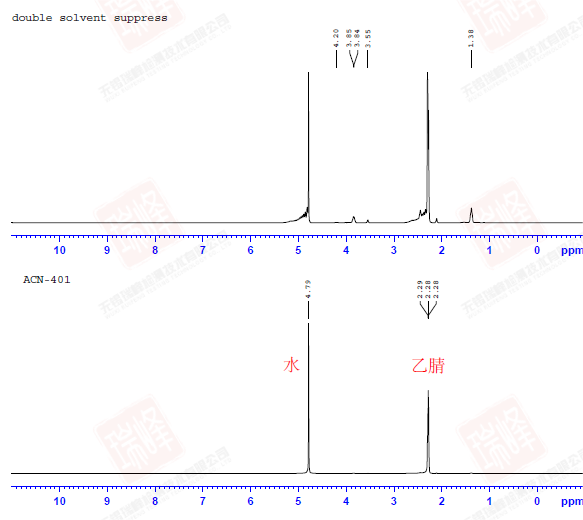
溶剂峰压制在核磁中是一个很重要的技术,尤其在做生物大分子或者LC-NMR联用时,溶剂信号甚至会是样品信号的10万倍,由于计算机的采样位数是固定的,大量的溶剂信号会几乎占满计算机的动态范围,而只留下很少的位数用来描述夹杂在噪音峰中的目标峰。上图中是回收乙腈的核磁谱图,在溶剂压制之前,几乎只能看到乙腈和水的信号;而在压制了两个溶剂峰之后,回收溶剂中的杂质峰渐渐显现。