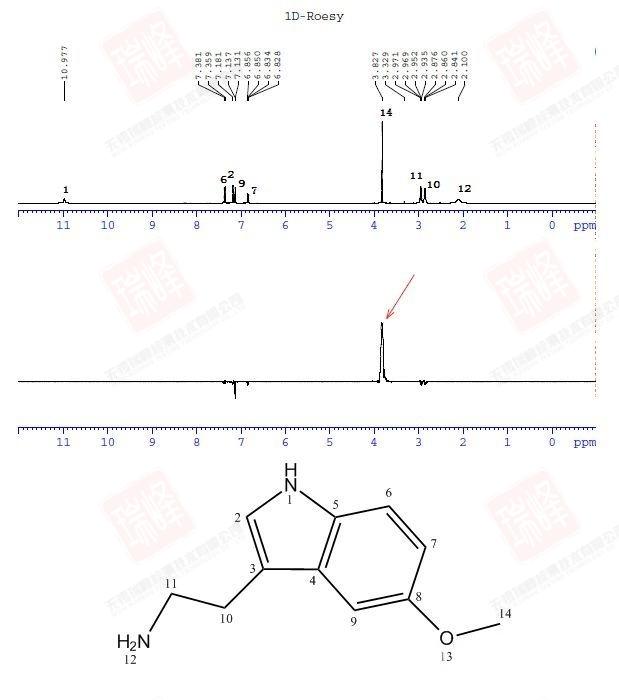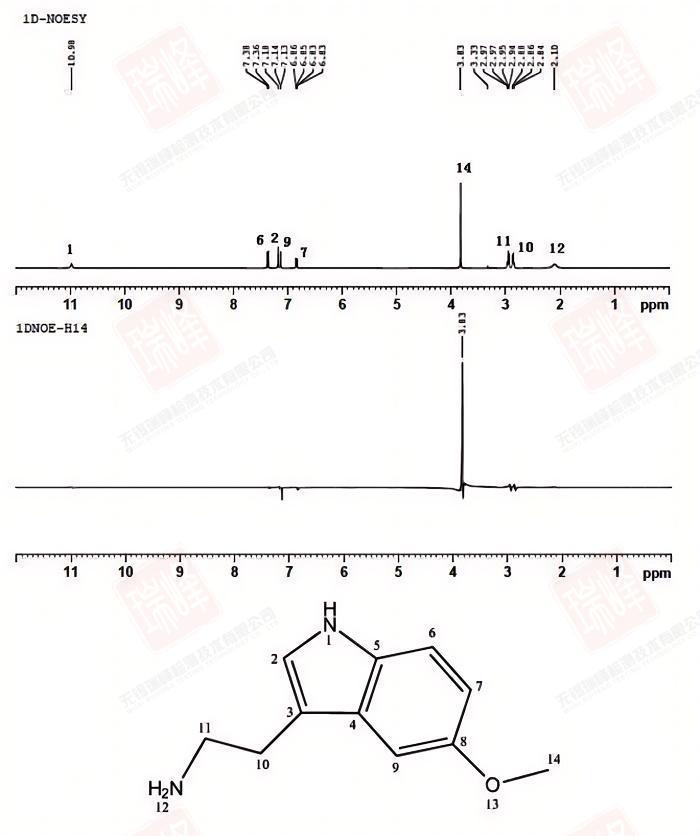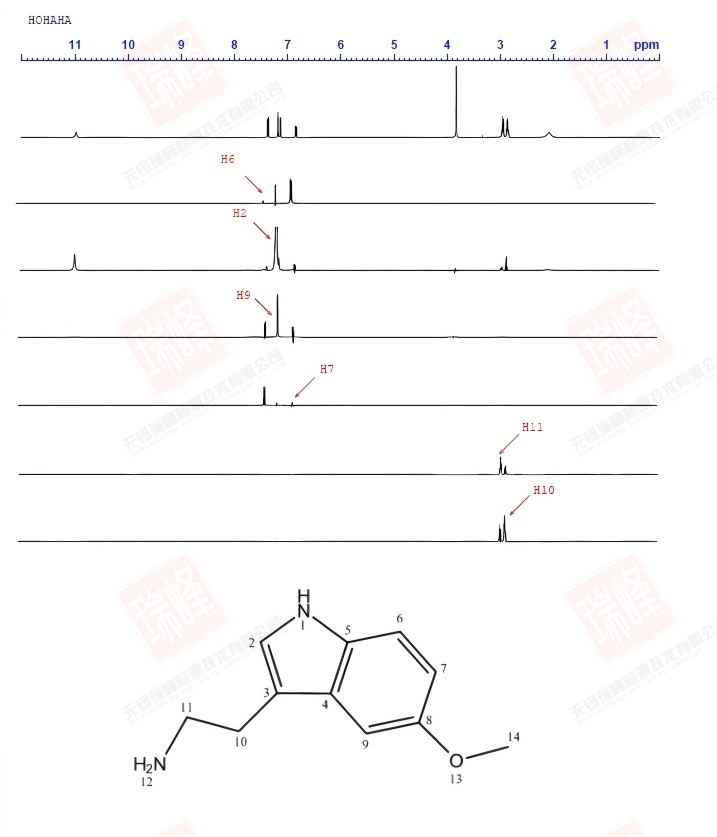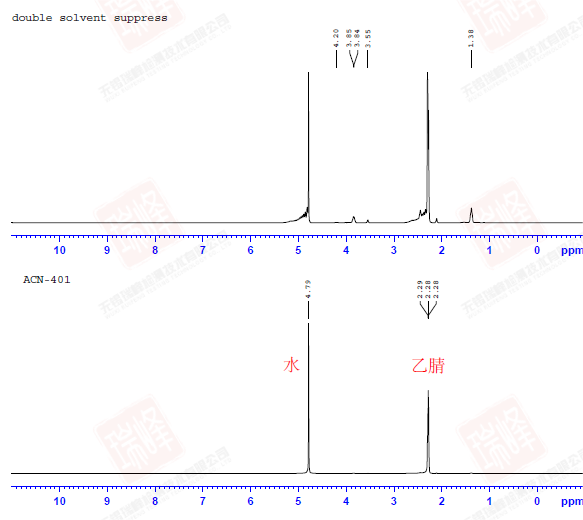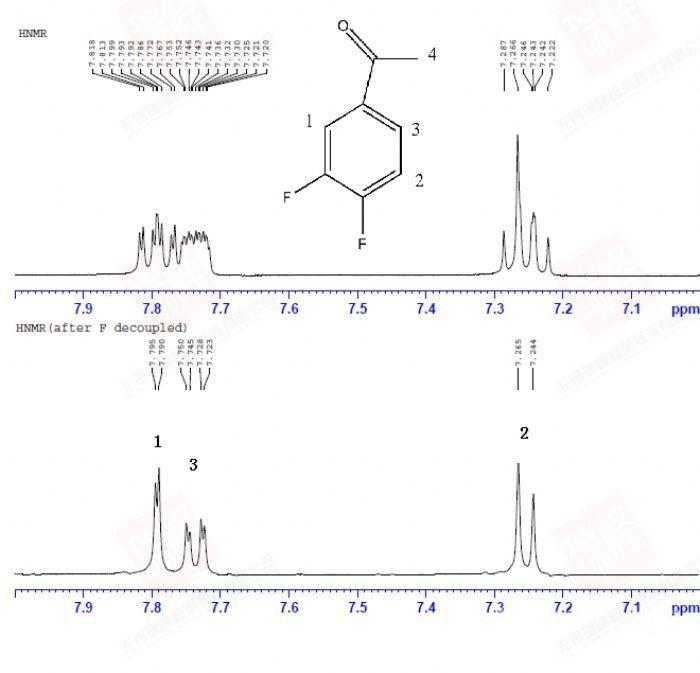
其实对判定结构而言,C核比H核更为重要,因为C是构成有机物的骨架,而H只是骨架上生出来的枝叶。遗憾的是,98.9%的12C由于自旋为0而不能被核磁检测到,而余下1.1%的13C因为旋磁比仅为H的1/4以及较长的弛豫时间导致核磁学家最终将H作为核磁实验中最重要的核。但C核绝没有被忽视。
之前已经提过,为了得到占C元素1.1%的13C信号,需要付出比H核多出数千倍的扫描时间。那么C-C相关信号呢?这个问题等价于分子中两个13C正好挨在一起的几率为多大——1.1%的平方。也就是说,10000个分子中只有一个分子某两个13C正好相邻,而要观察到这一个分子的CC相关,难度不仅在于如何“万里挑一”将这个很弱的信号采集出来,更在于如何“以一敌万”排除余下9999个分子大军的信号干扰。而描述这一问题的实验,科学家称之为INADEQUATE。
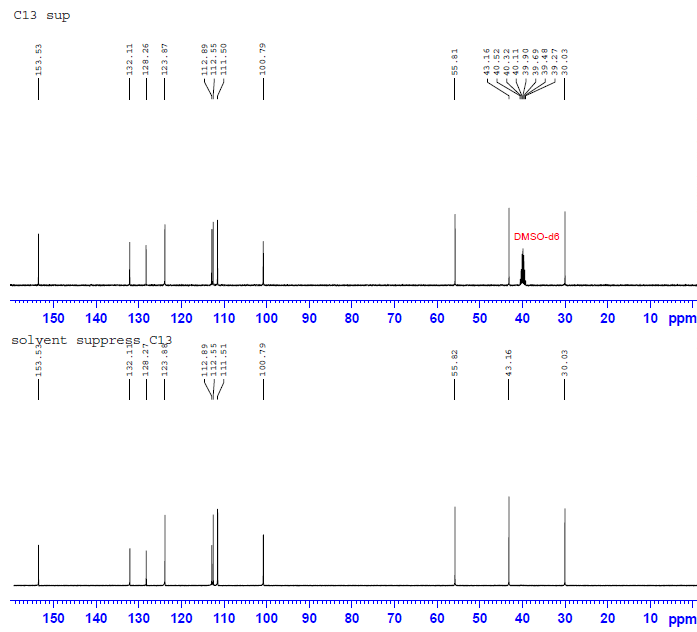
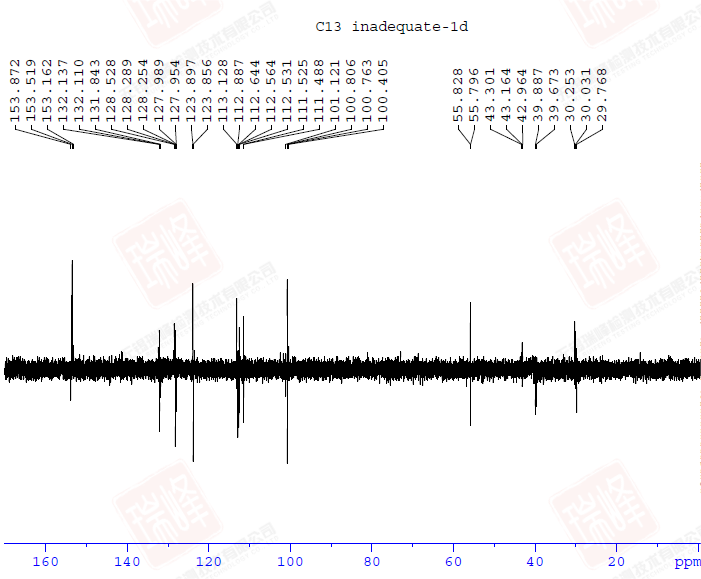
先给出这一结构的1D-INADEQUATE谱图。与2D相比,1D时间所花费的时间要少的多,但即使如此,这张谱图依然花费了我数小时的时间(还是在样品很浓的情况下)。由于脉冲演化的关系,1D-INADEQUATE实验中的信号常以反相位双重峰来呈现(其实稍微修改一下脉冲就可以实现相位同一化,但是我觉得这样反而不利于谱图解析),这是因为不管碳链多长,我们观察到的C-C相关通常就是一键相关(三个13C恰好相连的概率有多大?),而双重峰的波峰与波谷间距离是CC耦合常数,但遗憾的是,在1D-INADEQUATE中要分辨这一信号并不容易。
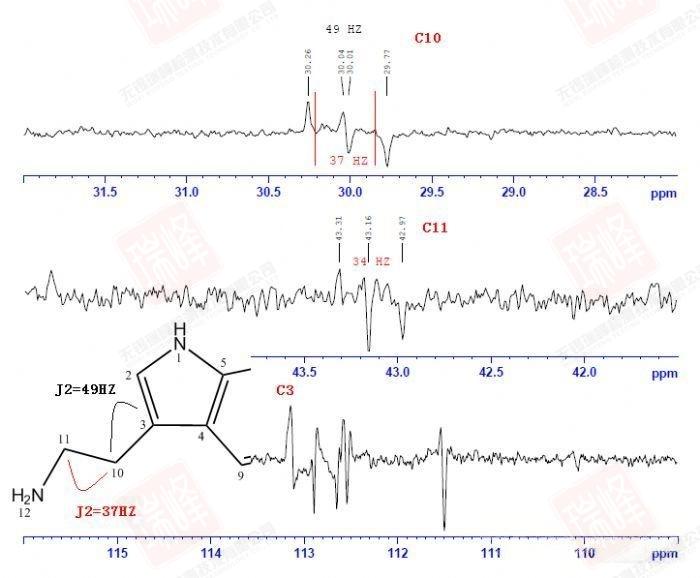
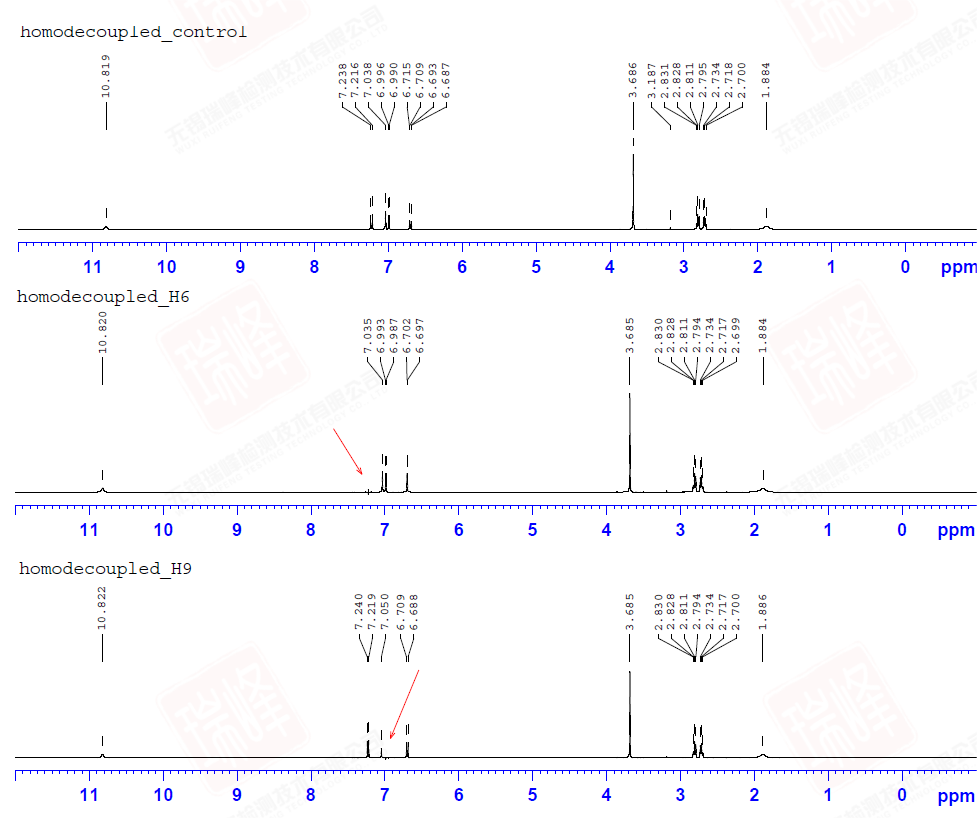
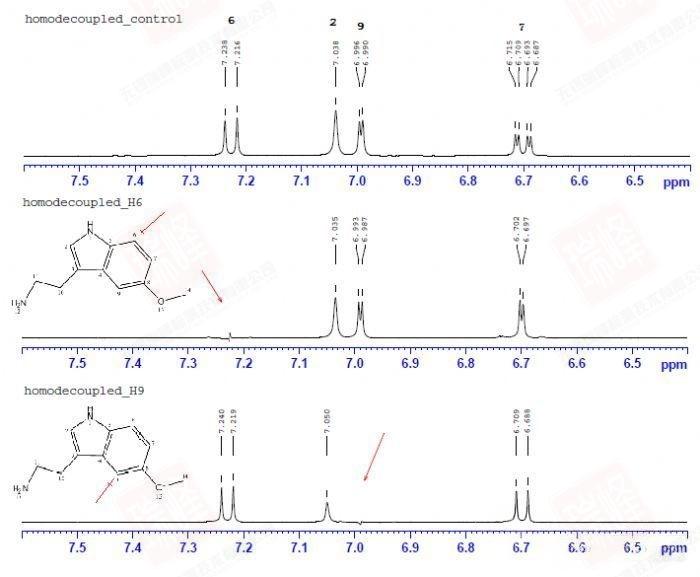
大家或许还记的之前在H归属中对于H10以及H11的归属曾经一度出现困难——HNMR中14N核四极矩造成的展宽以及H-N HMBC中相关信号的强弱都支持将高场H定为H11,但HMBC,COSY都明确给出了与之相反的结论。其实从C-C角度上来看这个问题会简单很多——C11仅受C10耦合,而C10却要受到C11和C3的耦合作用。图一中C10能明显读出49Hz的耦合作用,而红线标出的37Hz的反相位峰显得很弱,需要注意的是,在两个反相位双重峰中间位置出现的峰并不是三重峰,而是没有压制完全的13C单峰。图二给出的C11峰信号较弱,这可能是与之直接相连的14N所引起,34Hz的耦合值暗示着C10与C11的耦合关系。C3由于与C6与C7相互重叠,耦合变得难以辨认,使这一实验在解决C归属问题上不够严密,而我之后讲到的选择性1D-INADEQUATE将解决这个问题。